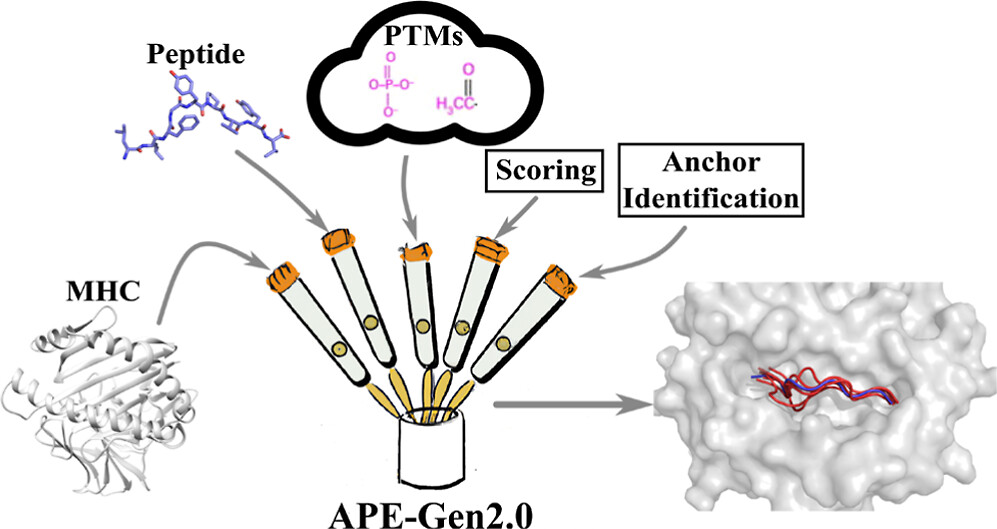
The APE-Gen2.0 tool. APE-Gen2.0 comprises different processes that increase pMHC structural modeling accuracy, expand the pMHC structural modeling repertoire, and make the tool easier to use by the community.
Overview
APE-Gen2.0 is a new pMHC structural modeling tool. Compared to it's predecessor, as well as other pMHC structural modeling tools in the literature, it exhibits better performance on pMHC structural modeling accuracy. Moreover, it expands the pMHC modeling repertoire to cases that we were previously unable to model. Such cases include peptides exhibiting post-translational modifications, such as phosphorylation and citrullination, among others. Other cases deal with peptides that exhibit non-canonical peptide geometries in the MHC binding cleft, such as peptides exhibiting N-terminus/C-terminus extensions. Lastly, all of these cases, and more, are able to be modeled through just very few clicks through this webserver.
Algorithm
APE-Gen2.0 is able to achieve state-of-the-art performance over its predecessor through very specific changes and contributions. Such changes can be seen in the flowchart on the left, and are enumerated here in more detail:
Anchor identification module: The anchor identification module, given a peptide sequence and an MHC allotype, is able to predict the anchor configuration of the peptide in the MHC cleft with great accuracy. Predictions also extend to non-canonical configurations also. As a result, the appropriate pMHC template is fetched that can allow modeling of non-canonical cases if need be.
Concurrent usage of sample and threading: The predecessor of APE-Gen2.0, as well as some other few pMHC structural modeling approaches in the literature use sampling and scoring padarigms for pMHC structural modeling. Other tools emphasize on pMHC modeling through a given structural template, like homology modeling, among others. APE-Gen2.0 uses boht of these paradigms concurrently. We find that this allows for more a complete sampling of the pMHC conformational space, and resulted in state-of-the-art performance
Post-translational modifications: APE-Gen2.0 is the first method that can rapidly model peptides exhibiting post-translational modifications. This is possible through adopting pyTMS, a post-translational modification modeling tool. Contrary to previous offerings in the literature, modeling of post-translational modifications happens within minutes.
A web interface: We now offer a pMHC modeling interface that allows pMHC modeling with a few clicks throgh this web server. Local installation of APE-Gen2.0 is still possible, however, the web server usage is recommended for users not wanting to install APE-Gen2.0 locally.